Neonatal encephalopathy, Hypoxic Ischemic Encephalopathy, and Subsequent Cerebral Palsy: Etiology, Pathology and Prevention.
George Malcolm Morley, MB ChB FACOG
Summary:
The clinical definition of neonatal encephalopathy (NE) is that used by Cowan et al., [1] hypoxic ischemic encephalopathy (HIE) is a defined radiological diagnostic finding, and cerebral palsy, (CP) is a broad neurological diagnosis of permanent CNS dysfunction. This paper defines the etiology and prevention of cerebral palsy that follows NE and HIE; the main reference for this paper is Cowan’s [1] paper.
Cowan concludes (from substantial evidence) that events in the immediate perinatal period are most important in neonatal brain injury. Analysis of Cowan’s criteria for the diagnosis of NE defines those events - every child that developed NE had been exposed to some degree of perinatal asphyxia/hypoxia, and every child that developed NE was hypovolemic. There is little evidence that hypoxia, per se, unless extreme, results in brain damage; all evidence points to hypovolemia as the primary perinatal pathogenic insult that causes NE.
The sequence of pathologies, NE / HIE / CP, occurs in newborns that have been subjected during birth to cord compression (partial occlusion of the umbilical vein) of sufficient degree to produce hypovolemia in the fetus and engorgement of the placenta; the sequence, NE / HIE / CP, is initiated by immediate clamping of the umbilical cord at birth (ICC) that finalizes and intensifies the hypovolemia in such a cord-compressed neonate. ICC prevents placental transfusion that could correct the neonatal hypovolemia. In the hypovolemic, ICC neonate, the transfer of neonatal blood volume to the lungs following the first breath results in marked systemic hypovolemia and hypovolemic shock. Loss of blood pressure causes loss of brain perfusion and neuron necrosis occurs in the most metabolically active areas of the brain. MRI scanning will later confirm areas of edema and decreased perfusion (neuron necrosis.) In less acute cases, gradual cardiac failure (from low blood pressure and lack of cardiac perfusion) may result in gradual damage to the more metabolically active areas of the brain.
Primate studies on totally asphyxiated neonates confirm that loss of blood pressure is a major factor in producing brain damage. Hypovolemic shock in the slightly/moderately asphyxiated human neonate is the obvious and most plausible genesis for NE / HIE and CP.
As “there is no evidence that brain damage occurs before birth,” the sequence of NE / HIE / CP should be avoidable by resuscitating neonates with the placental circulation intact. This allows full (and immediate) placental transfusion from the engorged placenta and corrects the primary pathology.
A Deductive analysis of Dr. Cowan’s Paper [1]:
The aim of the study [1] was to test the hypothesis that neonatal encephalopathy, (NE) early neonatal seizures (ENS) or both result from early neonatal insults. The study strongly indicates otherwise, that immediate perinatal events/insults result in NE and ENS. Analysis of the immediate perinatal events that are listed as defining NE indicates that two perinatal insults combine to form the primary cause of NE and the subsequent CP.
The authors use a strict clinical definition of NE: Abnormal tone, feeding difficulties and altered alertness, plus three of the following five additional criteria:
- Late FHR decelerations or meconium staining.
- Delayed respiration.
- Arterial cord blood pH less than 7.1
- Apgar scores less than 7 at 5 minutes.
- Multi-organ failure.
A second group (ENS) consisted of infants who had seizures within 72 hours of birth, but did not meet the criteria for NE.
The Immediate Cord Clamping Factor (ICC)
The perinatal professions do not recognize ICC as a perinatal insult, despite it being repeatedly defined as such over the past 200 years:
- “ … very injurious to the child”. [2]
- “The cord should not be cut until the pulsations have ceased.” [3]
- “[ICC] should be avoided … in certain unfavorable conditions the consequences may be fatal.” [4]
- “ICC can result in hypotension, hypovolemia and anemia …” [5]
In the additional defining criteria for NE, #1 and #2 indicate fetal distress, mandating ICC for emergency removal of the neonate to the resuscitation table; #3 mandates ICC cord clamping, [6] and indicates that nearly all infants had ICC for cord pH studies regardless of what the cord pH results were. An Apgar score >7 at five minutes, #4, indicates a much lower score at birth, resulting in ICC to move the child to resuscitation. One may therefore conclude that all children with NE [1] had their cords clamped immediately and that not one received a full placental transfusion.
The first three mandatory criteria for NE are assessed by the neonatologist and are post-partum neurological phenomena. Despite some separation in time of these signs and symptoms from immediate perinatal events, all are related to low Apgar assessments of tone and reflexes. A pink, breathing newborn with a heart rate above 100 bpm (absence of asphyxia) that is limp and without reflexes scores Apgar six; this Apgar combination is impossible as breathing is a product of reflexes. Thus the first three mandatory criteria combined with additional criterium #4 imply some impairment of neurological function soon after birth; however, they do not confirm physical brain injury during the first five minutes after birth.
The Perinatal Insults
Additional criteria #2 and #3 are direct implications of intrapartum asphyxia being associated with NE, and #1 (late decelerations) is diagnostic for antepartum compression of the umbilical cord that impedes the fetal oxygen supply, asphyxiating the fetus. Some degree of intrapartum asphyxia (hypoxia and acidosis) is therefore an essential component in the clinical definition of NE. As 80% of babies thus defined had radiological “evidence of acutely evolving brain lesions” post partum, (neuron necrosis as opposed to suppressed function) this essential component, asphyxia/hypoxia, would appear to be reasonably valid in relation to causation. However, the authors also supply evidence that intrapartum asphyxia is not the primary or the only causal agent in the pathogenesis of NE, and that it intrapartum asphyxia may be incidental to brain damage.
- “There is no evidence that brain damage occurs before birth.”
- “There is often an absence of evidence of
severe antepartum asphyxia in infants with neonatal encephalopathy, and
conversely,”
- “Many infants who do have signs of fetal
distress and asphyxia do not develop neurological sequelae.”
- “Infants scanned within 24 h. of birth
might have had normal scans, even though scans obtained later in the first
week showed severe injury.”
Thus clinical NE,
when correlated with radiological hypoxic ischemic encephalopathy (HIE),
appears and progresses after the child is born, and during that time,
the resuscitated child is seldom severely asphyxiated / hypoxic. The multiple organs that fail (#5 in the
authors’ definition) while HIE progresses, grow and develop in utero with a
blood supply that has a relatively low oxygen partial pressure due to mixing of
umbilical venous blood with vena caval blood.
Thus these organs, including the brain, are relatively resistant to
damage from moderate hypoxia. In the
otherwise normal newborn, a minor to moderate degree of pulmonary asphyxia
alone should not cause HIE or multiple organ failure, and in group 2, (ENS),
ischemia, infarction and hemorrhage were the predominant pathologies; hypoxia
was not mentioned. Most acute tissue
infarcts (caused by arterial occlusion) become hemorrhagic due to bleeding into
necrotic areas from collateral circulation.
This brings into question the importance or even the validity of the
word “hypoxic” in the term “hypoxic ischemic encephalopathy;”
“More than 90% of
term infants with NE, seizures or both had evidence of perinatally acquired
insults, [1] and there was a very low rate of established brain injury before
birth.” The conclusions from all the
above findings are that the perinatal insult that results in NE / ENS / HIE /
seizures and multi-organ failure
- Occurs at, or very near to, the time of
birth.
- Has pathogenic effects persisting after
birth for at least five minutes.
- It is not pure asphyxia (hypoxia and/or
acidosis.)
The authors [1]
identify such an insult: “Nelson and
colleagues have noted that a tight nuchal cord at delivery was seen
significantly more often in infants with neonatal encephalopathy than in
controls, which lends further support to the explanation that events late in
labor are important in neonatal encephalopathy.”
The authors [1] also
include such an insult in their definition criteria – “late decelerations on
fetal monitoring.” Late decelerations
on the fetal monitor result in rapid obstetrical delivery of the child, vaginally
or abdominally – they thus occur late in labor and are diagnostic of cord
compression; this is the most common cause of “fetal distress.” A tight nuchal cord, a firm true knot in the
cord, a prolapsed cord, occult or vaginal, a cord firmly wrapped around a limb,
or a cord compressed between any body part and the uterine wall (most commonly
seen in association with oligo-hydramnios (and possibly relieved by
amnio-infusion)) will result in late decelerations of the fetal heart rate if
the uterine contraction increases the pressure on the cord.
Late decelerations
are produced by contraction of the uterus increasing the cord compression and
impeding the flow of oxygenated cord venous blood to the fetus. The resultant hypoxia and decreased venous
return to the fetal heart result in fetal bradycardia that starts after the
uterine contraction appears on the monitor and recovers during uterine
diastole.
Cord compression thus results in asphyxia, and, as concluded above, asphyxia is not the primary perinatal insult that results in NE; however, cord compression, by depriving the child of oxygen also deprives the child of blood volume that carries the oxygen. [7] Cord compression acts as a venous tourniquet, impairing fetal cord venous blood flow while umbilical arteries engorge the placenta with fetal blood volume. The cord-compressed child is born asphyxiated, and hypovolemic.
The routine treatment of such a child is to doubly clamp the cord immediately to obtain a blood sample for pH determination, and to transfer the child rapidly to resuscitation. Although the time of cord clamping is not documented, [1] from analysis of the additional criteria for the definition of NE, noted above, one must conclude that all neonates in the study had immediate cord clamping (ICC) at birth for these purposes. A large portion of each child’s natural blood volume was therefore discarded with the engorged placenta and / or sent to the laboratory.
At physiological (normal) delivery, when the cord vessels are allowed to close physiologically, the normal child receives a large placental blood transfusion (30% to 50% increase in blood volume [5]) that is generated by gravity or by uterine contraction. [8] Placental circulation (respiration, preventing asphyxia) normally continues after birth until pulmonary function occurs. [4] In the case of the cord-compressed fetus, born asphyxiated and hypovolemic, ICC, by abolishing placental oxygenation and placental transfusion, imposes complete asphyxia and severe hypovolemia on the apneic neonate. Despite adequate pulmonary resuscitation, the child may lack sufficient blood volume to establish adequate pulmonary circulation, resulting in hypovolemic shock and 5-minute Apgar scores below 7. Multi-organ failure in hypovolemic shock is typified by oliguria / anuria and progressive kidney damage if blood volume is not promptly and adequately restored. Correct cord management at birth relieves cord compression [3] and restores physiology.
The absence of late
decelerations does not necessarily preclude cord compression as a cause of
fetal distress. Descent of the neck and
shoulders with uterine contraction, pulling the cord tighter, produces the
typical late deceleration seen in a tight nuchal cord. If fetal movement tightens a cord loop
around a limb, the deceleration pattern seen in the following figure may be the
only sign of cord compression. It often
is the initial incident before late decelerations are subsequently seen.
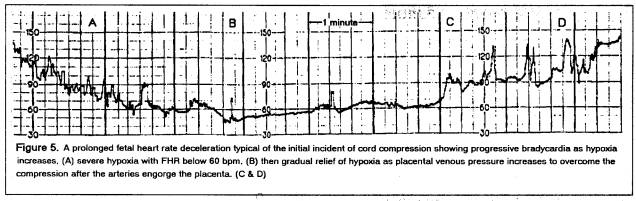
Such an event usually results in nurses rapidly changing the patient’s position (to knee-chest.) In the above case, the patient slept through the episode that was discovered 20 minutes later when the FHR pattern was normal apart from a slight suggestion of late FHR decelerations. Occult cord prolapse was found at C-section; the child was pallid at birth but had recovered to Apgar ten at 5 minutes after which the cord was then clamped.
After such a deceleration, the child is hypovolemic but not hypoxic, as the high placental venous pressure ensures adequate cord venous return. However, high hydrostatic pressure in the placental capillaries results in fluid loss from the fetus; if the cord compression is not reversed, severe fetal dehydration (that accentuates the hypovolemia) will develop.
The child that is born extremely depressed following deep, prolonged, late decelerations from a tight cord around the neck (Apgar 1 – heart rate 60) is not blue-black in color; it is ashen white with some patches of deeply cyanotic skin. Generalized reflexive vaso-constriction selectively limits effective circulation to the heart, brain and placenta, and if the placental blood volume is removed (by ICC) before the lungs are ventilated, establishment of the pulmonary circulation with filling of the pulmonary vasculature after ventilation (that relaxes pulmonary arterioles) may result in marked decrease in systemic cardiac output. [4] As blood pressure is a product of cardiac output and peripheral resistance, ventilation of the immediately clamped, hypovolemic neonate may, by filling the lungs with blood, collapse the systemic blood pressure.
The kidneys and other neonatal tissues (including the brain) are relatively immune to hypoxic damage as long as perfusion is adequate. However, loss of perfusion (low/zero blood pressure) results in rapid infarction and necrosis regardless of blood oxygenation. Inadequate perfusion results in gradual infarction.
Primate studies by Windle [9] and Myers using ICC with delayed pulmonary respiration [10] produced “birth asphyxia” brain damage and cerebral palsy in monkeys. The autopsied monkey brains showed lesions that were virtually identical to the lesions in autopsied human brains in this present study – that also involved ICC and delayed respiration. [1] These primates were not subjected to antepartum cord-compression asphyxia and were not born hypovolemic or asphyxiated; all the “asphyxia” and placental blood loss were imposed after birth, and permanent brain lesions were produced within several minutes or more of total “asphyxia” followed by pulmonary resuscitation.
Infarction / tissue necrosis in the “asphyxiated” neonatal primate brain and in human NE occurs in areas of active metabolism – areas that are very dependent on adequate perfusion. Inadequate perfusion of itself entails hypoxia. In the “totally asphyxiated” monkeys subjected to ICC, the blood pressure (perfusion force) fell within minutes to near zero. [[10], Fig 1] A few more minutes of near zero blood pressure (inadequate tissue perfusion) resulted in permanent brain damage that was confirmed months or years later by autopsy on monkeys with cerebral palsy. Windle and Myers attributed the brain damage to asphyxia; however, the brain damage was very probably primarily caused by a short interval of virtual cardiac arrest (zero blood pressure) induced by the asphyxia. Resuscitation (reversal of asphyxia) in these primate studies restored cardiac function and blood pressure; it could not restore neuron necrosis.
The methods used to produce asphyxia in these two primate studies differed. Myers used ICC and a rubber glove over the neonate’s head up to the point of resuscitation. Windle delivered the placenta and membranes intact with the fetus inside, and ruptured the membranes after a recorded time to effect resuscitation; he did not record blood pressure or the time of cord clamping. In both studies the extent of brain damage increased with the period of asphyxiation. When the rubber glove [10] was not used, the neonate recovered (spontaneous respiration.) One must conclude, therefore, that the neuro-pathogenic hypotension in Myers’ cases was due to decreased cardiac output produced by anoxia, and that the loss of placental blood volume due to ICC in a normal monkey will not, of itself, produce brain damage. Peltonen states that the normal child survives clamping before the first breath without harm, but that in unfavorable circumstances, the consequences may be fatal. [4]
There were no continuous blood pressure recordings on the neonates in Cowan’s [1] study during the minutes after birth. The NE babies were already hypoxic and hypovolemic when subjected to ICC; however, the MRI findings of ischemia indicate loss of, or inadequate tissue perfusion in areas of tissue necrosis and infarction. Multi-organ failure occurs in hypovolemic shock regardless of oxygenation, again indicating that hypotension and inadequate organ perfusion were present in these neonates and are, therefore, the logical cause of the brain ischemia and infarction. As in the primate studies, several minutes of extreme hypotension and hypoperfusion may have occurred immediately after birth, or a more prolonged period of moderate hypotension – inadequate perfusion – may have produced the same result – neuron necrosis. Again, necrosis would be expected to occur in areas of very active metabolism rather than having an arterial distribution. The acute, severe hypotensive insult would appear to be the most plausible cause of neuron necrosis and correlates with the low five-minute Apgar score. An early MRI scan done after some recovery in blood pressure and tissue perfusion could well be negative, while a later scan would show the accumulated edema and ischemia in the necrotic areas.
The importance of blood pressure and the relative unimportance of oxygenation in preserving brain tissue are illustrated in one of Myer’s primate tracings in which total fetal anoxia was induced by maternal anoxia; the fetus was delivered by cesarean section after marked deceleration of the heart rate had occurred. Resuscitation was done with the placental circulation intact, and the cord was clamped long after pulmonary oxygenation was established. No significant fall in blood pressure occurred during the asphyxia, (i.e. brain perfusion was not impaired) and the neonate recovered fully. [[10], Fig 16]
In a minority of ENS infants in the study, [1] focal infarction was noted and, if arterial in distribution, would indicate vasospasm as a causative factor. In these cases, if a vasoconstrictor drug had been used to maintain blood pressure, the infarct may have been iatrogenic.
Discussion:
It is impossible to define pathogenic factors (insults) and their effects on an organ when the physiological factors necessary for the life and health of that organ are not defined and understood. In the case of life support for the newborn brain during the third stage of labor, an extremely complex switch occurs from placental life support to independent neonatal life support systems, yet physicians are poorly educated regarding that physiology; in one particular aspect they are grossly misinformed. The health and life of cerebral neurons is acutely dependent on adequate perfusion of nervous tissue with blood that contains adequate amounts of nutrients and oxygen. In the fetus, cerebral blood flow is generated by two cardiac ventricles while nutrients and oxygen are supplied by the placenta; in the newborn, the left ventricle alone is the motive force for brain perfusion, the right ventricle and the lungs generate oxygen supply, and nutrients are obtained mainly by perfusion of the liver, and later the gut. The fetal / neonatal life-support switch involves radical anatomical and physiological changes, and to maintain neuron integrity during the switch (adequate brain perfusion with suitable blood), placental function is normally maintained until the newborn life support systems are functioning. [4]
A major factor in establishing and activating the newborn’s life support systems and in maintaining adequate cerebral perfusion during birth is the transfer of a large volume of blood from the placenta to the newborn – placental transfusion. [8] After the newborn has achieved a (physiological) blood volume that is optimal for survival, and after the lungs are functioning, the cord vessels close reflexively and the placenta is infarcted. Most physicians and midwives have (and have been taught) the misconception that placental transfusion is a pathological event that causes over-transfusion, polycythemia, hyperviscosity and jaundice. Thus immediate cord clamping produces a newborn with a low blood volume that is mistakenly deemed to be “normal”; it is the un-clamped child that achieves a normal blood volume; the ICC child is hypovolemic. [5]
Consequently, most physicians have never (consciously) seen a newborn with a physiological blood volume, and most radiologists have never read an MRI done on a newborn that has optimal cerebral perfusion following physiological cord closure / placental transfusion. Establishment of the physiological norm for neonatal brain perfusion is an absolute needed before inadequate (pathological) perfusion can be objectively diagnosed. Scans presently deemed to be normal might thus differ markedly from the actual physiological norm.
A major accomplishment of Cowan’s research [1] is the refutation of the recent claim by the task force of The American College of Obstetricians and Gynecologists (ACOG) that “neonatal encephalopathy and cerebral palsy (CP) are largely not caused by labor and delivery events.” Cowan’s data clearly indicate perinatal insults as the cause of CP.
Another great value of this study [1] is the concise and specific clinical definition of a very complex syndrome that has a great variety of ultimate outcomes. Without this defined foundation, the syndrome remains a muddle of seemingly contradictory factors. This confusion is illustrated in a dissertation on hypoxic-ischemic brain injury [11] that explains the origin of the term “newborn encephalopathy” (NE) as medico-legal terminology alternative to HIE contrived to relieve obstetricians from unfair blame for poor outcomes in asphyxiated babies; however, the description in this dissertation of the pathophysiology of HIE illustrates the lack of comprehension of birth physiology, especially in relation to newborn blood volume and cardiac output.
The resistance of fetal tissues to hypoxia is noted, [11] and the effect of severe hypoxia on myocardial function (cardiac failure) is also noted as a causal factor in cerebral hypoperfusion. Severe prenatal or postnatal hemorrhage is mentioned as a cause of circulatory failure; however, blood loss into the excised placenta is not recognized as hemorrhage. Rearrangement of cardiac output that shunts blood from liver, kidneys, lungs and gut to the heart and brain is attributed to undefined “hypoxic-ischemic insults” and “This initial reaction to systemic hypoxia and ischemia is one of lowering the heart rate and increasing the blood pressure to maintain a near normal cardiac output.” [11] This statement contradicts all known concepts of cardiac physiology, and the concept of ICC causing hypoxia and hypovolemia (ischemia) is not even recognized.
Complete fetal asphyxia produced by occluding the umbilical cord results in a reduction of about 50% in the heart rate (120 bpm to 60 bpm – bradycardia); to maintain “near normal” cardiac output would require doubling of the stroke volume – a 100% increase in ventricular volume prior to each systole – and this does not occur. Hypoxia (and/or a fall in central venous pressure or systemic blood pressure) causes extreme vaso-constriction in all body parts except the heart, brain and lungs (or placenta); this results in increased peripheral resistance, an increase (or maintenance) of blood pressure, and eventually a marked decrease in venous return to the heart. The end result is bradycardia [Fig, 5] with a very marked reduction in cardiac output but with preservation of (blood pressure) perfusion through the heart, brain and lungs (if functioning) or the placenta. Continued lack of oxygen results in myocardial failure, decrease in cardiac output and blood pressure, with eventual inadequate perfusion of heart and brain. Cardiac function essentially ceases and neuron necrosis begins; the cardiac function can be restored with correction of oxygenation and blood volume, the neuron necrosis cannot. [10]
In the case of pure hypoxia, oxygenation should prevent this sequence, and if instituted before deficient perfusion (from heart failure) has caused brain damage, it should result in full recovery. Thus pure hypoxia / anoxia may suppress brain function completely, but until the hypoxia / anoxia results in myocardial failure and loss of blood pressure, (loss of brain perfusion) the integrity of nervous tissue is usually maintained. Abruptio placenta creates such an instance, and mimics Windle’s primate studies. Pulmonary oxygenation with the placenta still attached (restoration of blood volume) should restore normality in such cases unless hypoxia has already caused heart failure and brain damage.
If, however, circulatory failure is primarily due to pure hypovolemia, (inadequate venous return to the heart) and peripheral vasoconstriction cannot support adequate blood pressure to perfuse heart and brain, neuron necrosis will occur and progress rapidly in brain areas of active metabolism. In such a case, oxygenation becomes a minor factor in neuron necrosis as oxygen delivery to heart and brain is inadequate. In the ICC neonate, a combination of hypoxia and hypovolemia is involved; in the cases presented in the present study, [1] all neonates were hypoxic and hypovolemic at birth, and they all received a severe hypoxic , hypovolemic insult a few seconds after they were born – immediate cord clamping. The cause of neuron necrosis in NE [1] is loss of tissue perfusion due to inadequate blood pressure. Inadequate blood pressure results from cardiac failure caused by loss of blood volume. Hypoxia alone, unless it is severe and prolonged, is not a significant causal factor in NE.
Preemies were excluded from this study. The area of the preemie’s brain that is by far the most metabolically active is the germinal matrix that ceases to function around 36 – 37 weeks gestation. Preemies are routinely subjected to ICC, and the most common hypoxic, ischemic hemorrhagic infarct seen in preemies is germinal matrix / intra – ventricular hemorrhagic infarction.
Of the five additional defining criteria for NE, [1] only #1 (late decelerations) is an antepartum event; it indicates fetal asphyxia and hypovolemia. Delayed respiration, #2, implies early cord clamping (before respiration is established) for transfer to resuscitation; ICC causes loss of placental transfusion and thus hypovolemia. [5] Arterial cord blood pH less than 7.1, #3, (ICC [6]) is an intra-partum procedure and indicates hypovolemia due to loss of placental transfusion. Apgar score less than 7 at five minutes, #4, indicates depression due to a factor other than hypoxia; with resuscitation, the lungs are well oxygenated after five minutes, and hypovolemia is a plausible explanation for the depression. Multi-organ failure, #5, occurs post partum; when placed in context with multiple hypovolemic incidents and the resistance of the organs to hypoxia, its only plausible genesis is hypovolemic shock. Thus all five additional criteria needed for a diagnosis of NE strongly imply that hypovolemia is a major factor in that diagnosis, and hypovolemia becomes a mandatory factor in the causation of NE as herein defined.
Several criteria need to be documented to confirm hypovolemia as the primary cause of NE, namely, the time of cord clamping, the neonate’s blood pressure at the time of Apgar scorings, urine output and the central venous pressure. Corresponding normal values on neonates whose umbilical cords close physiologically also need to be established. The use and timing of blood volume expanders and /or blood transfusion on the neonates in this study [1] and the later development of anemia would also be informative.
To reproduce NE in a primate study, a cord tourniquet, gradually inflated to 20 – 30 mms Hg should be applied to the undelivered fetus at C-section. When the “distress” has stabilized, (the placenta is engorged at 30mms Hg cord venous pressure, restoring cord venous flow) the cord should be clamped and the baby delivered and resuscitated. The newborn will have sustained maximal intra-placental blood loss and the resultant hypovolemia should result in development of NE.
Conclusion:
Therefore NE, as defined in the study [1] and as diagnosed radiologically, occurs after birth from hypo-perfusion of the brain resulting from hypoxic, hypovolemic shock caused by ICC performed on an already hypovolemic, hypoxic, cord-compressed neonate. It follows that NE, as defined in the study [1], should be preventable to a very large extent by not clamping the umbilical cord.[3][2]
Thus two perinatal insults combine to produce HIE. Cord compression during birth causes hypoxia and hypovolemia, upon which ICC imposes hypoxia and extreme hypovolemia immediately post partum; deficient perfusion of the brain results in hypoxic ischemic encephalopathy – infarction of metabolically active areas of the child’s brain. In other words, intrapartum cord compression shifts a large portion of the child’s blood volume to the placenta, and ICC ensures that the child is ischemic after birth, and prone to ischemic complications. The cord clamp is the pathogenic instrument.
The authors [1] define NE as a limp child, unable to suckle, slow to respond, “much weaker than it ought to be,” that deteriorates into multi organ failure and brain damage after a large portion of its blood is left in the placenta following a “very injurious” procedure. [2] When Hammersmith and Wilhelmina Hospitals have delivered 351 depressed, asphyxiated newborns and resuscitated them with their placental circulations intact according to Erasmus Darwin’s and Charles Meigs’ instructions, [2][3] and have prevented NE in 90+% of these neonates, they will have completed the perfect control series for this superb study.[1]
References:
- Frances Cowan et al. Origin and Timing of Brain Lesions in Term Infants with Neonatal Encephalopathy. The Lancet, Vol.361, Issue 9359,1 March, 2003 pages 736-742
- Darwin E. Zoonomia 1801 Vol III page 321
- Meigs C. Professor of Obstetrics and Diseases of Women and Children, Jefferson Medical College. A Philadelphia Practice of Midwifery, 1842
- Peltonen T. Placental Transfusion, Advantage - Disadvantage. Eur J Pediatr. 1981;137:141-146
- Linderkamp O. Placental transfusion: determinants and effects. Clinics in Perinatology 1982;9:559-592
- ACOG Committee Opinion Number 138 - April 1994, published in the International Journal of Gynaecology and Obstetrics 45:303-304 [54], reaffirmed 2000, and listed as current in OBSTETRICS & GYNECOLOGY, February 2002,
- Cashmore j, Usher RH. Hypovolemia resulting from a tight nuchal cord at birth. Pediatr Res 1973;7:339
- Gunther M. The transfier of blood between the baby and the placenta in the minutes after birth. Lancet 1957;I:1277-1280.
- Windle WF (1969) Brain damage by asphyxia at birth. Scientific American 221(#4): 76‑84.
10. Myers RE (1972) Two patterns of perinatal brain damage and their conditions of occurrence. American Journal of Obstetrics and Gynecology 112:246-276.30.
- Hypoxic-Ischemic Brain Injury in Newborn Authored by Marcio Sotero de Menezes, MD, Assistant Professor, Department of Neurology, Division of Pediatric Neurology, Children's Hospital of Seattle, University of Washington, www.emedicine
- Morley. GM Neonatal Resuscitation: The life that failed. OBGYN.NET
NOTES:
Meyers:
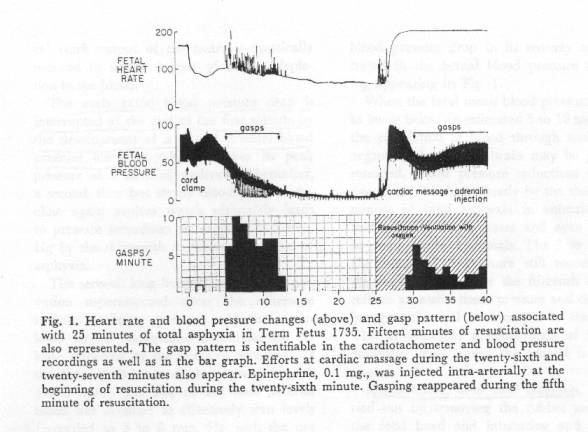
Myers, Fig.1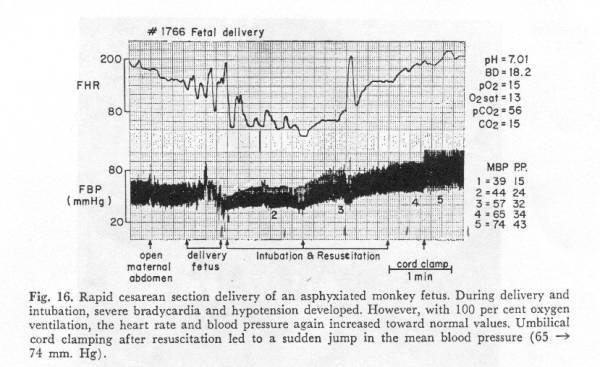
Myers, Fig 16.
Erasmus Darwin:
“Another thing very injurious to the child, is the tying and cutting of the navel string too soon; which should always be left till the child has not only repeatedly breathed but till all pulsation in the cord ceases. As otherwise the child is much weaker than it ought to be, a portion of the blood being left in the placenta, which ought to have been in the child.”
Erasmus Darwin, Zoonomia, 1801
Meigs: Regarding clamping a cord around the neck:
“The head is born: perhaps the cord is turned once, or even more than once around the child’s neck, which it encircles so closely as to strangulate it. Let the loop be loosened to enable it to be cast off over the head. … [or] by slipping it down over the shoulders. … If this seems impossible, it should be left alone; and in the great majority of cases, it will not prevent the birth from taking place, after which the cord may be cast off. … Should the child be detained by the tightness of the cord, as does rarely happen, … the funis may be cut … Under such a necessity as this, a due respect for one’s own reputation should induce him to explain, to the bystanders, the reasons which rendered so considerable a departure from the ordinary practice so indispensable. I have known an accoucheur’s capability called harshly into question upon this very point of practice. I have never felt it necessary to do it but once. … The cord should not be cut until the pulsations have ceased.”
Charles D
Meigs, M.D. Professor of Obstetrics and Diseases of Women and Children,
Jefferson Medical College. A
Philadelphia Practice of Midwifery, 1842
|
Author's Email: |
|
|
|
|
Editor's Email: |
|
|
eMedicine Journal, May 13 2002, Volume 3, Number 5
|
|
INTRODUCTION |
Section 2 of
10 |
Background: The terms hypoxic-ischemic encephalopathy (HIE) of
the newborn and perinatal asphyxia have been used synonymously in the past,
rather loosely. Clinical signs of HIE often are wrongfully considered to be
always the result of intrapartum asphyxia. This misconception has led to HIE
being considered a marker of perinatal obstetric mismanagement, leading to many
medicolegal problems. In reality, establishing a clear relationship between
perinatal brain injury and ischemia/hypoxemia is often difficult. The term
birth asphyxia is also imprecise, and its use is not recommended, because it
implies that intrapartum anoxia has occurred.
In the immediate newborn period, many
factors can produce neurological symptoms mimicking HIE, including prepartum
and postpartum ischemia/hypoxemia, genetic factors, metabolic disease, and
maternal and fetal drug use. Since establishing the relationship between
asphyxia and HIE is not always possible, the term newborn encephalopathy (NE) was
proposed as an alternative to remove the medicolegal implications of HIE.
Newborn encephalopathy is a clinically defined syndrome of disturbed
neurological function in full-term infants that attempts to correlate symptoms
in the neonatal period that have some relationship with neurological outcomes
in childhood. NE symptoms may or may not be linked causally to
hypoxemia/ischemia. Far from "fixing" the problem, the use of the
term NE just removes the unfair blame for poor neonatal outcomes from obstetric
practitioners.
The National Collaborative Perinatal Project
(NCPP), a prospective study of more than 50,000 pregnancies and 40,000 infants,
analyzed the features of NE and found that the following were associated with
increased morbidity on follow-up examinations: decreased activity after the
first day of life, need for incubator more than 3 days, feeding problems, poor
suck, and respiratory difficulties.
Other factors not mentioned in the NE
syndrome description have been associated with postneonatal morbidity such as
static motor deficits (cerebral palsy [CP]), mental retardation, and epilepsy.
These factors include neonatal seizures, low 10-minute Apgar scores, stupor,
and coma.
Pathophysiology:
General principles of perinatal ischemia
and hypoxemia
In fetal life, since the arterial partial
pressure of oxygen (PaO2) normally is quite low, hypoxic-ischemic
disturbances are primarily a consequence of hypoperfusion. In spite of this,
severe hypoxemia can occur, leading to myocardial dysfunction with subsequent
cerebral hypoperfusion or loss of cerebrovascular autoregulation. This cerebral
hypoperfusion in turn may lead to neuronal ischemia.
Primary perinatal hypoxemia
In utero hypoxemia is usually the result of
placental insufficiency, and infants who have experienced in utero hypoxemia
often have significant respiratory or cardiac failure after birth. On the other
hand, postnatal hypoxemia is the result of either respiratory or cardiac
insufficiency, alone or in combination. Primary perinatal hypoxemia can derange
the already fragile cerebrovascular autoregulation of the neonatal brain (see Pressure-passive cerebral
circulation). This may explain why severe surfactant deficiency syndrome is
associated with certain patterns of neuronal damage, such as periventricular
leukomalacia.
Perinatal ischemia
Severe cardiac contractile dysfunction due
to either major cardiac malformations or severe hypoxemia leads to cerebral
hypoperfusion and loss of cerebrovascular regulation. Circulatory insufficiency
can result from severe prenatal or postnatal hemorrhage or neonatal sepsis. In
response to hypoxic-ischemic insults, circulatory rearrangement of the cardiac
output occurs, with shunting of blood flow away from the liver, kidneys, gut,
lungs, and skeletal muscle into the heart, brain, and adrenals of the infant.
This shunting explains the coexistence of liver and kidney failure in cases of
severe HIE-NE. This initial reaction to systemic hypoxia and ischemia in the
newborn is one of lowering the heart rate and increasing blood pressure to
maintain a near-normal cardiac output.
With progression of the hypoxic-ischemic
state, the heart rate, blood pressure, and cardiac output decrease
significantly; systemic metabolic acidosis increases, in part because of
production of lactic acid.
Pressure-passive cerebral circulation
In the neonatal period, autoregulation of
cerebral circulation is impaired. This autoregulation is further impaired by
both hypoxemia and hypercarbia, which leaves the cerebral circulation in what
is termed a pressure-passive state. In this state, cerebral perfusion changes
as the intravascular pressure changes. The immature cerebral arteries have a
limited ability to adapt to hypotensive episodes. This may be especially
significant in the genesis of the parasagittal pattern of cerebral injury that
is seen following hypotension. The other extreme of arterial pressure range
also may be problematic, since the decreased upper limit of autoregulation may
lead to an increased risk of periventricular and intraventricular hemorrhages.
Poor perfusion of the depths of the sulci
In the neonatal period, the depths of the
sulci are relatively underperfused, creating a zone of increased susceptibility
to hypotensive insults. This may lead to gyri that resemble mushrooms, owing to
atrophy of their base near the deeper part of the sulci.
…
George Malcolm Morley, MB ChB FACOG
10252 E. Johnson Road
Northport, MI 49670
Copyright G. M. Morley, MB ChB FACOG, August 5, 2003